La hipercolesterolemia familiar (HF) es una enfermedad hereditaria que se expresa desde el nacimiento, y que cursa con un aumento en las concentraciones plasmáticas de colesterol, principalmente del colesterol transportado por las lipoproteínas de baja densidad (c-LDL).
Es un trastorno muy frecuente y se estima que al menos 1 de cada 250 personas en la población general presenta HF. En España, se calculan en unas 200.000 las personas con HF.
La importancia de su diagnóstico precoz radica en el elevado riesgo de presentar un Infarto de Miocardio (IM) u otra enfermedad ateroesclerótica vascular en edades tempranas de la vida. La enfermedad cardiovascular se manifiesta en más del 50% de los pacientes con HF antes de los 55 años de edad.

¿Qué causa la HF y como se hereda?
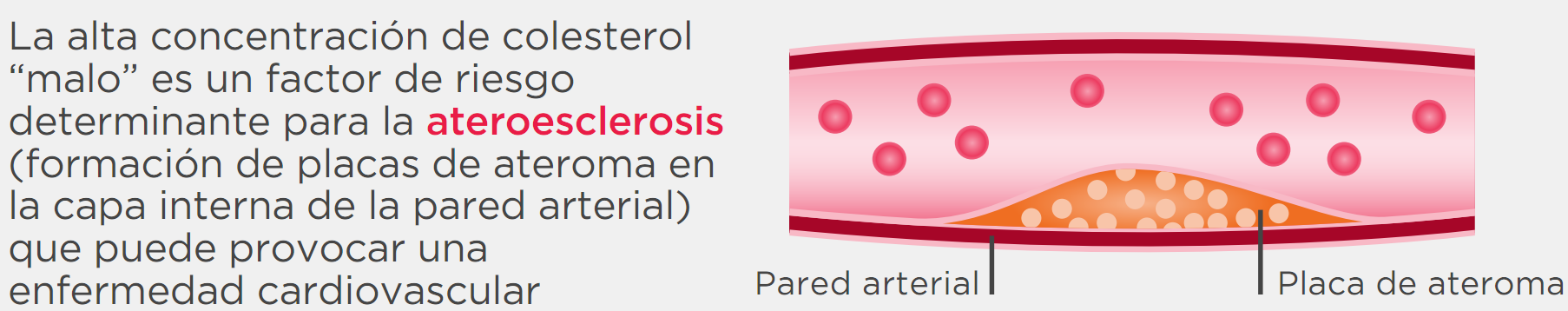
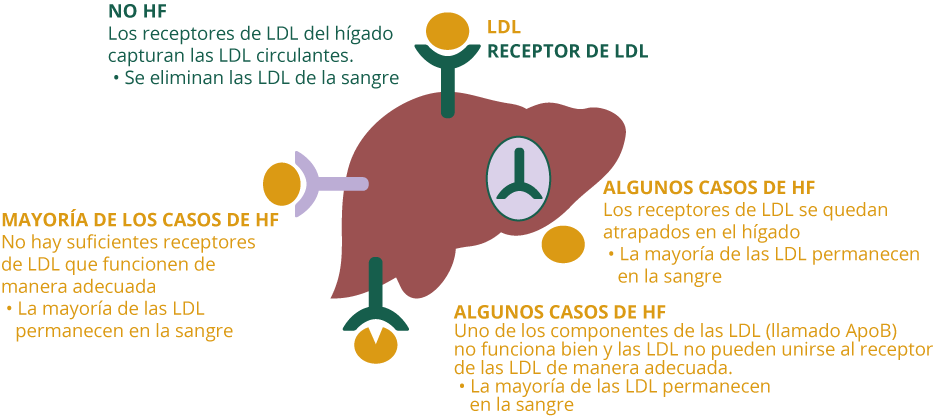
El defecto principal se produce por una mutación en el gen que codifica el receptor de las LDL (rLDL), que son los encargados de eliminar el colesterol de la sangre a nivel hepático. Al disponer de una menor cantidad de receptores, ya sea parcial o total, el colesterol LDL aumenta considerablemente en la sangre, favoreciendo su depósito en las arterias y el desarrollo de una placa que puede estrechar la luz de las arterias, lo que produce la ateroesclerosis.
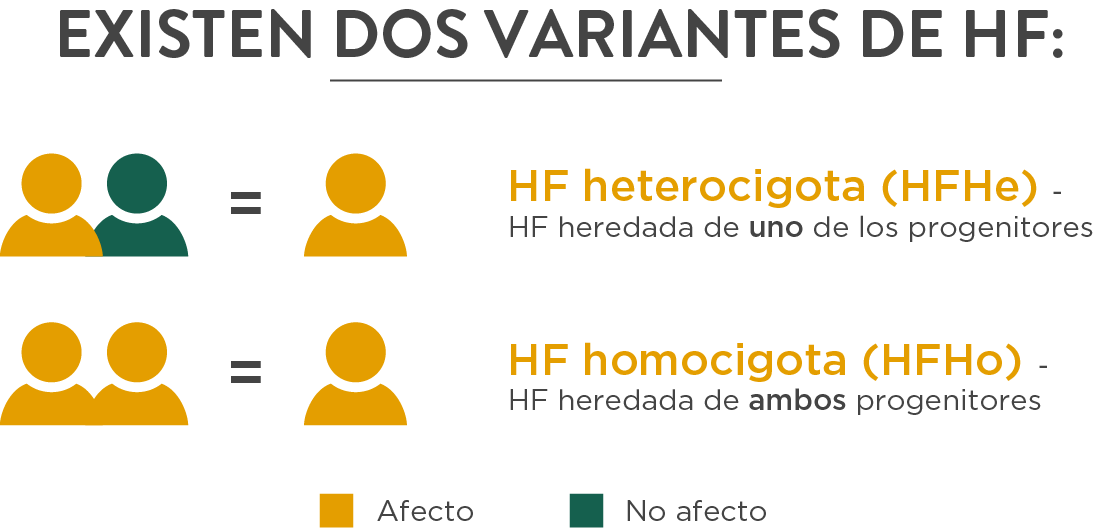
Por su mecanismo de transmisión, se reconocen dos variantes:
- Heterocigota: uno de los alelos tiene una mutación en el gen y el otro es normal. En este caso, el paciente tiene el 50% de la dotación de receptores-LDL normofuncionantes, y el resto están ausentes (mutaciones de alelo nulo) o no funcionan adecuadamente (mutaciones de alelo defectuoso).
- Homocigota: ambos alelos están defectuosos (el del padre y la madre), lo que produce una ausencia prácticamente total de receptores LDL.
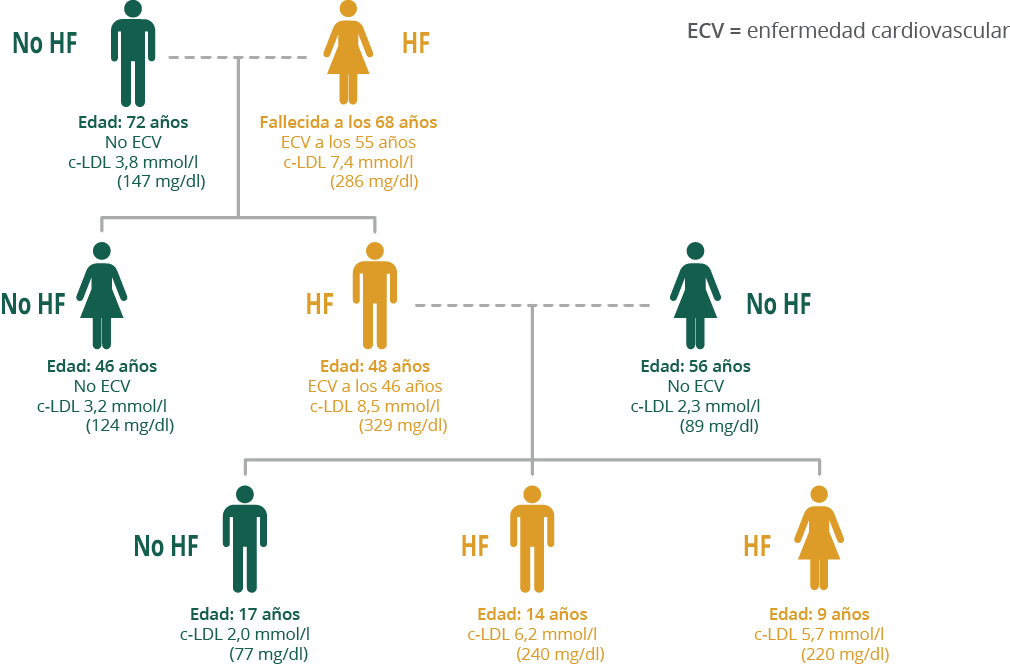
Una persona afecta de HF, tiene el 50% de probabilidades de transmitir el gen anormal a sus descendientes, hijos e hijas, y un 50% de traspasar la información genética correcta. Por lo tanto, aproximadamente la mitad de los miembros de una familia heredarán la HF. Si un niño o adulto, hijo de un paciente con HF, tiene niveles normales de colesterol, es muy probable que haya heredado el gen normal, y por tanto no desarrollará la enfermedad ni la transmitirá a su descendencia. Algunos estudios han demostrado que puede existir hasta un 8% de personas portadoras de una mutación con niveles normales de colesterol. En este caso pueden transmitir el gen defectuoso a su descendencia. Por eso, es muy importante la realización del diagnóstico genético.
En pocas palabras
La HF es una enfermedad genética muy frecuente que se transmite de padres a hijos. Las personas afectadas tienen niveles de colesterol alto, debido habitualmente a que el colesterol no es eliminado correctamente en el hígado por una escasez de receptores.
Actualmente, se conocen más de 1.600 mutaciones diferentes a lo largo de todo el gen del rLDL en individuos con hipercolesterolemia familiar procedentes de diversas poblaciones a nivel mundial. Cuando una población se encuentra aislada geográfica o culturalmente, o cuando una gran proporción de personas se encuentra emparentada por descender de antecesores comunes a causa de la migración, podrán existir una o muy pocas mutaciones en un grupo amplio de población. Sin embargo, en otros países como España, donde la población es más heterogénea desde el punto de vista genético, el número de mutaciones suele ser mucho mayor. Hasta la fecha se han reconocido más de 450 mutaciones distintas causantes de HF en España.
La variabilidad en la expresión clínica de la HF en cuanto a las concentraciones de colesterol y el desarrollo de enfermedad cardiovascular, y parte de la respuesta al tratamiento hipolipemiante depende en parte de la mutación del gen del rLDL.
En cifras: Hipercolesterolemia Familiar
- 1 de cada 250 personas en la población general presenta HF (200.000 personas en España).
- La enfermedad cardiovascular se manifiesta en más del 50% de los pacientes con HF antes de los 55 años de edad.
- Una persona afecta de HF, tiene el 50% de probabilidades de transmitir el gen anormal a sus descendientes, hijos e hijas.
- Se conocen más de 1.600 mutaciones diferentes a lo largo de todo el gen del rLDL relacionadas con la HF.
Además de la Hipercolesterolemia Familiar, cuyo defecto genético está en el gen del rLDL, existen otras causas de hipercolesterolemia familiar que se expresan de forma similar (Tabla 3).
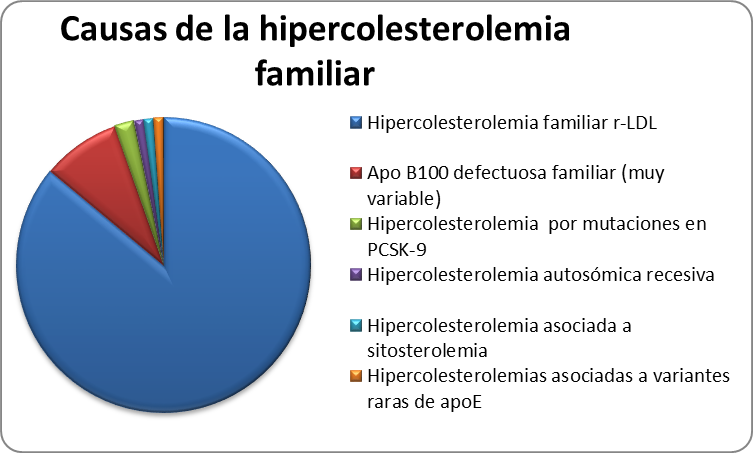
Tabla 3. Frecuencia de hipercolesterolemias familiares autonómicas dominante
Estas son:
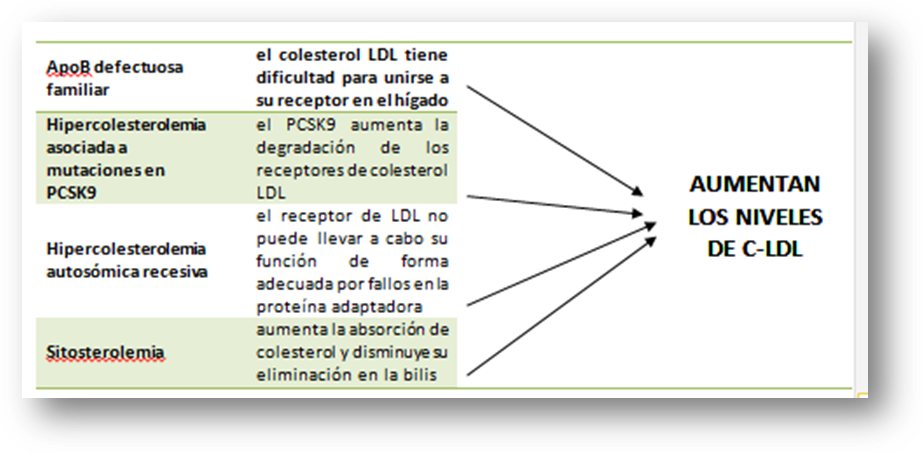
Apo B defectuosa familiar
La apolipoproteína B (ApoB100) es la única proteína de las LDL y la responsable de unirse al rLDL en el hígado. Los defectos moleculares en el gen de la ApoB100 producen una proteína con una capacidad reducida de unirse al rLDL, por tanto los niveles de c-LDL serán similares a los observados en la HF. Hasta la fecha, se han descrito cuatro mutaciones en este gen asociadas a hipercolesterolemia, la más frecuente, sustituye la Arginina de la posición 3500 por Glutamina y se conoce con el nombre de ApoB3500.
Las características clínicas de esta hipercolesterolemia son prácticamente idénticas a las causadas por defectos en el rLDL, y se conoce como Apo B defectuosa familiar (BDF). Ambas entidades solo pueden distinguirse por el análisis genético.
La mutación apo B3500 es frecuente en países centroeuropeos y poco frecuente en el norte y sur de Europa. Se cree que esta mutación tiene una antigüedad de más de 6000 años y es de origen Celta. En España representa aproximadamente el 4%-5% de la población con hipercolesterolemia autosómica dominante. Sin embargo, es una causa frecuente de hipercolesterolemia en la población gallega, probablemente por su origen Celta.
Hipercolesterolemia asociada a mutaciones en PCSK9
Varios estudios han encontrado familias en las cuales a pesar de presentar un fenotipo de HF, el defecto responsable de la hipercolesterolemia no está localizado ni en el gen del rLDL ni en el gen de la apo B100. En el 2003 se describieron algunas mutaciones en el gen de la Proproteína convertasa subtilisina/kexina tipo 9 (PCSK9) que producen un aumento en la función de esta proteína y se asocia a hipercolesterolemia severa. Esta proteína tiene un papel clave en la degradación de los rLDL hepáticos, y por tanto contribuye a regular los niveles plasmáticos de c-LDL. En este sentido, favorece la degradación intracelular de los rLDL, disminuyendo su reciclaje hacia la membrana del hepatocito, lo que ocasiona una reducción del número de receptores y por tanto un aumento en los niveles de colesterol-LDL. En España, aunque se han encontrado casos esporádicos de HF producidas por mutaciones en este gen, es muy infrecuente.
Hipercolesterolemia autosómica recesiva (ARH)
La ARH se produce por mutaciones en la proteína adaptadora del rLDL y tiene un patrón de herencia autosómico recesivo. Los síntomas y niveles de c-LDL son similares a los observados en pacientes con HF homocigota. Ambos progenitores son heterocigotos obligados para el defecto molecular en el gen que codifica la proteína adaptadora, pero por regla general, tienen niveles normales de c-LDL a diferencia de los pacientes con HF homocigota. Este tipo de hipercolesterolemias se asocian también con enfermedad coronaria prematura y xantomas tendinosos.
Sitosterolemia (hipercolesterolemia familiar pseudohomocigota)
Es un trastorno autosómico recesivo muy poco frecuente, que se caracteriza por aumento en la absorción intestinal de esteroles vegetales, entre los que se encuentra el sitosterol y el colesterol de la dieta, además de eliminar menos colesterol a través de la bilis. Los síntomas clínicos son similares a los de la HF homocigota con una elevada incidencia de enfermedad aterosclerótica prematura y xantomatosis. Por regla general este tipo de pacientes responden bien a la restricción de los esteroles vegetales de la dieta y al tratamiento con ezetimiba (un inhibidor de la absorción intestinal del colesterol). En España se han descrito solamente unos pocos casos.
En pocas palabras
Comentarios desactivados