Además de la Hipercolesterolemia Familiar, cuyo defecto genético está en el gen del rLDL, existen otras causas de hipercolesterolemia familiar que se expresan de forma similar (Tabla 3).
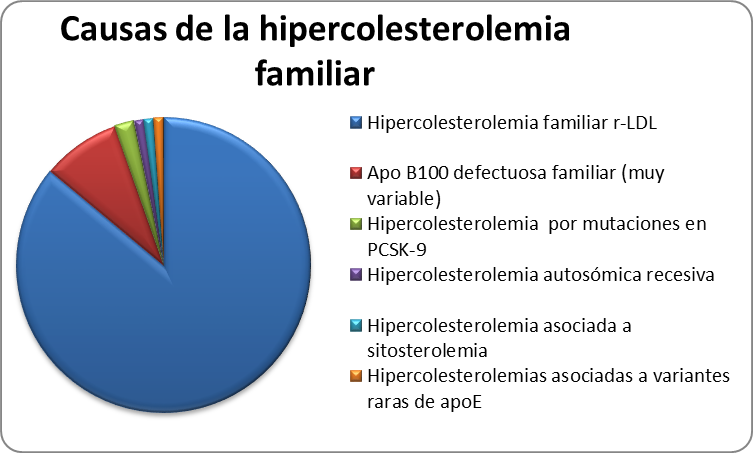
Tabla 3. Frecuencia de hipercolesterolemias familiares autonómicas dominante
Estas son:
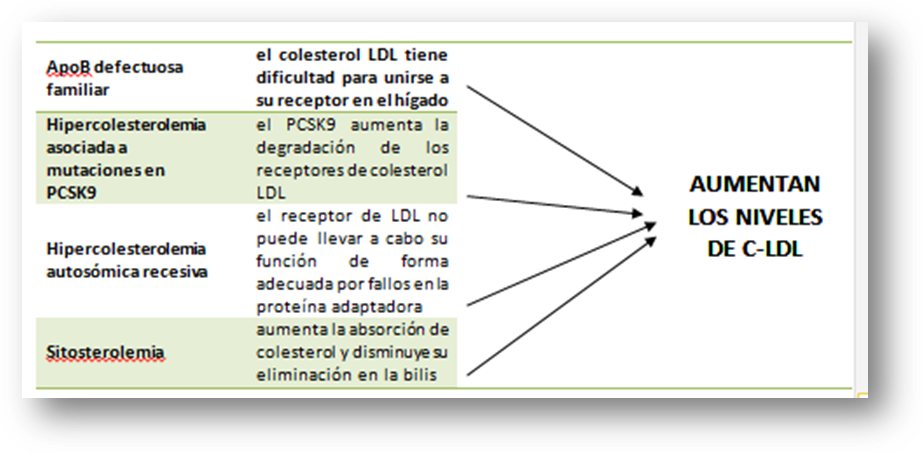
Apo B defectuosa familiar
La apolipoproteína B (ApoB100) es la única proteína de las LDL y la responsable de unirse al rLDL en el hígado. Los defectos moleculares en el gen de la ApoB100 producen una proteína con una capacidad reducida de unirse al rLDL, por tanto los niveles de c-LDL serán similares a los observados en la HF. Hasta la fecha, se han descrito cuatro mutaciones en este gen asociadas a hipercolesterolemia, la más frecuente, sustituye la Arginina de la posición 3500 por Glutamina y se conoce con el nombre de ApoB3500.
Las características clínicas de esta hipercolesterolemia son prácticamente idénticas a las causadas por defectos en el rLDL, y se conoce como Apo B defectuosa familiar (BDF). Ambas entidades solo pueden distinguirse por el análisis genético.
La mutación apo B3500 es frecuente en países centroeuropeos y poco frecuente en el norte y sur de Europa. Se cree que esta mutación tiene una antigüedad de más de 6000 años y es de origen Celta. En España representa aproximadamente el 4%-5% de la población con hipercolesterolemia autosómica dominante. Sin embargo, es una causa frecuente de hipercolesterolemia en la población gallega, probablemente por su origen Celta.
Hipercolesterolemia asociada a mutaciones en PCSK9
Varios estudios han encontrado familias en las cuales a pesar de presentar un fenotipo de HF, el defecto responsable de la hipercolesterolemia no está localizado ni en el gen del rLDL ni en el gen de la apo B100. En el 2003 se describieron algunas mutaciones en el gen de la Proproteína convertasa subtilisina/kexina tipo 9 (PCSK9) que producen un aumento en la función de esta proteína y se asocia a hipercolesterolemia severa. Esta proteína tiene un papel clave en la degradación de los rLDL hepáticos, y por tanto contribuye a regular los niveles plasmáticos de c-LDL. En este sentido, favorece la degradación intracelular de los rLDL, disminuyendo su reciclaje hacia la membrana del hepatocito, lo que ocasiona una reducción del número de receptores y por tanto un aumento en los niveles de colesterol-LDL. En España, aunque se han encontrado casos esporádicos de HF producidas por mutaciones en este gen, es muy infrecuente.
Hipercolesterolemia autosómica recesiva (ARH)
La ARH se produce por mutaciones en la proteína adaptadora del rLDL y tiene un patrón de herencia autosómico recesivo. Los síntomas y niveles de c-LDL son similares a los observados en pacientes con HF homocigota. Ambos progenitores son heterocigotos obligados para el defecto molecular en el gen que codifica la proteína adaptadora, pero por regla general, tienen niveles normales de c-LDL a diferencia de los pacientes con HF homocigota. Este tipo de hipercolesterolemias se asocian también con enfermedad coronaria prematura y xantomas tendinosos.
Sitosterolemia (hipercolesterolemia familiar pseudohomocigota)
Es un trastorno autosómico recesivo muy poco frecuente, que se caracteriza por aumento en la absorción intestinal de esteroles vegetales, entre los que se encuentra el sitosterol y el colesterol de la dieta, además de eliminar menos colesterol a través de la bilis. Los síntomas clínicos son similares a los de la HF homocigota con una elevada incidencia de enfermedad aterosclerótica prematura y xantomatosis. Por regla general este tipo de pacientes responden bien a la restricción de los esteroles vegetales de la dieta y al tratamiento con ezetimiba (un inhibidor de la absorción intestinal del colesterol). En España se han descrito solamente unos pocos casos.
En pocas palabras
Comentarios desactivados